
Recently, I was one of 16 recipients of a 10 mill. DKK grant (1.3 mill. EUR) from the VILLUM foundation under their Young Investigator Program (YIP). The program is unique in Denmark and offers young scientists an opportunity to build a research group on their own terms. The foundation is working on the premise of the founder of who famously said:
“One experiment is better than a thousand expert opinions”
Villum Kann Rasmussen
Hence, they simply support good experiments and trust that the researchers will come up with great solutions, if the foundation interfere as little as possible. This means as little as possible administration and flexible funding if new opportunities arise during the project. While this sounds almost too good to be true, previous grantees have all said that it actually works this way!
So, how do I plan to spend 10 mill DKK (1.5 mill. EUR)?
Microbial communities underpin all processes in the environment and have direct impact on human health. Despite their importance, only a tiny fraction of the millions of different microbes is known. This is mainly due to the immense difficulties of cultivating microbes from natural systems in the laboratory. This discrepancy is also known as the “microbial dark matter”.
For any microbe, the genome is the blueprint of its physiological properties. Having this in hand, it is possible to reconstruct its potential metabolism and establish hypotheses for evolution, function and ecology. Furthermore, it provides a foundation for further validating its function through a variety of in situ methods. However, genomes are extremely difficult to obtain from the microbial dark matter.
Currently, multiple metagenomes combined with bioinformatic approaches, is used to retrieve individual genomes from complex samples (see e.g. our paper from 2013). This has let to numerous fundamental discoveries, including the discovery of bacteria cable of complete ammonia oxidation (Comammox, see here and here), which radically change our view of the global nitrogen cycle and granted us the “Danish research result of the year, 2015”.
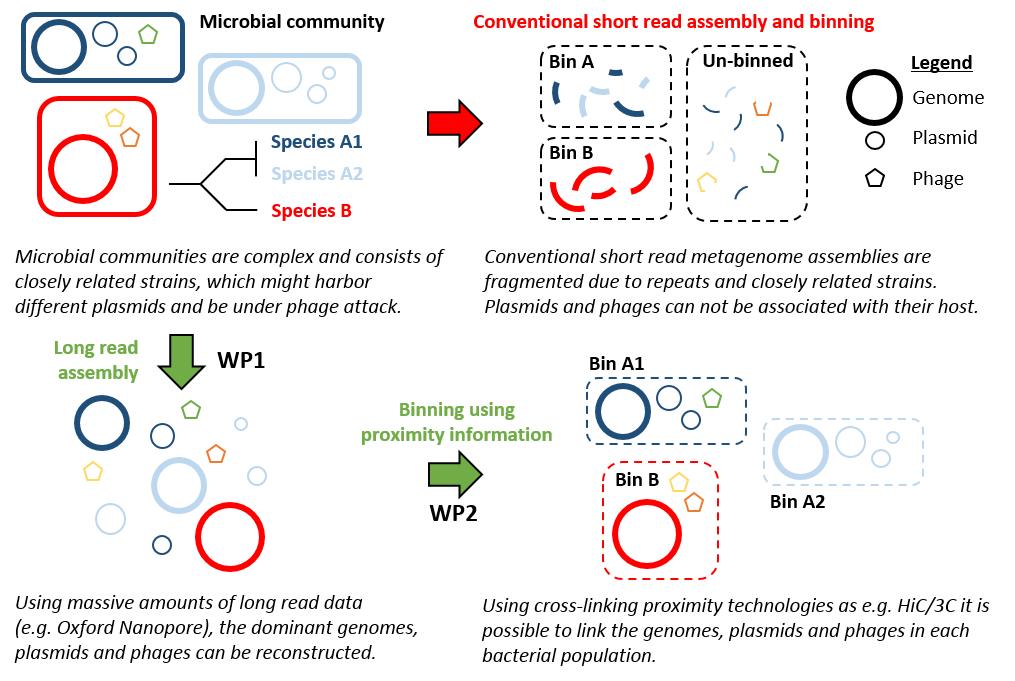
However, we are still far from realizing the full potential of metagenomics to retrieve genomes. Mainly due to the complexity of nature, where multiple closely related strains co-exists, which renders the current approaches useless.
Using the VILLUM YIP grant we want to use cutting-edge DNA sequencing related techniques to enable access to all genomes despite strain-complexity, link genomes, plasmids and phages, and enable direct measurements of in situ bacterial activity. The ability to readily obtain activity measurements of any bacteria, in any microbial ecosystem, will radically change microbial ecology and environmental biotechnology.
Obtaining complete bacterial genomes
Retrieving individual bacterial genomes from complex microbial communities can be compared to mixing hundreds of puzzles with millions of pieces, all containing different shades of blue sky. However, one way to circumvent the problem with closely related strains, is to use bigger pieces of DNA to assemble the genomes. The current standard approach is to use short read sequencing (Illumina; approx. 2 x 250 bp). However, the rapid development within long-read DNA sequencing, means that it is possible to start to experiment and envision how this is going to be solved.
The newest technology to the long read market is the Oxford Nanopore. It has successfully been used to generate complete genomes from pure cultures and we have used it for metagenomics of simple enrichment reactors to obtain the first complete Comammox genome. We have been early access testers of the MinION and are currently involved in the developer program. The improvement of the technology that has happened in the first half of 2016, means that the quality and throughput of the technology are now sufficient to attempt medium complexity metagenomes. Furthermore, we are one of the early customers to the high-throughput version of the MinION, the PromethION, which, in theory, would allow us to tackle even complex metagenomes.
Furthermore, while long-read DNA sequencing might enable closed bacterial chromosomes, they are still not associated directly with e.g. plasmids and phages. However, the last couple of years the several new methods have appeared, e.g. Hi-C and 3C, that utilize physical cross-linking of the DNA inside cells to generate sequencing libraries where proximity information can be retrieved. This information can then be used to infer which genetic elements were in close proximity, and thereby originated from the same bacterial cell. However, until now, the methods have only been used in microbial communities of limited complexity, but there is does not seem to be theoretical limits that would hinder the use of the methods, if complete genomes are available.
Measuring in situ replication rates using DNA sequencing
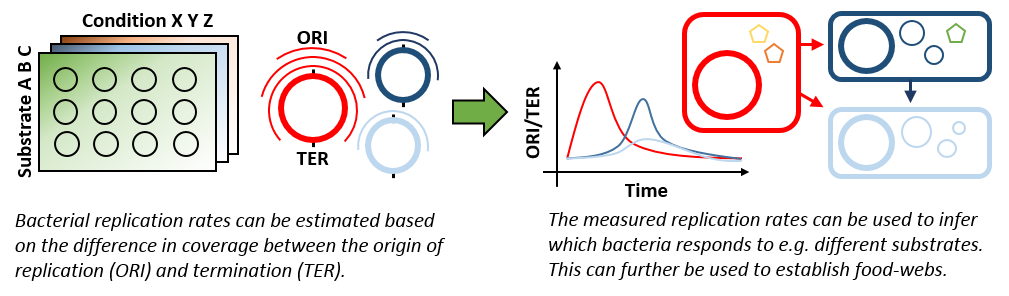
An exciting new possibility is that complete genomes enable measurements of bacterial replication rates directly from metagenomic data (see here and here). The method is very simple and based on the fact that the majority of bacteria starts replication at a single origin and then proceeds bi-directional. Hence, in an active dividing bacterial population there will be more DNA at the origin of replication than at the termini. This can be directly measured using DNA sequencing, as the number of reads is proportional to the amount of DNA in the original sample. Hence, by comparing the number of reads (coverage) at the origin to the termini, a measure of bacterial replication rate is obtained. This allows direct observations of individual bacterial response to stimuli in the environment, even in very complex environments as e.g. the human gut and with sub-hourly resolution. This type of information has been the dream of microbial ecologists since the field emerged over 100 years ago and will allow for countless new experiments within microbial ecology. Recently, the method has even been demonstrated to work with high-quality metagenome bins (see here). It is going to be interesting to further explore the potentials and limitations of the method using complete genomes at an unprecedented scale.
A few closing remarks
I am thrilled to have the next five years to explore how we can apply new DNA sequencing methods to understand the bacterial world and have the chance to build up a group of young scientists that share my excitement! If you think the project sounds great and either want to collaborate or work with us – then drop me an email!
Finally, I have to thank the people and mentors that made this possible. First of all my long-term mentor Per H. Nielsen; 6 years ago he introduced me to the world of microbial communities and throughput the years he have given me the freedom to pursue my own ideas – “freedom with responsibility” as we say in Danish. A leadership style that I very much try to adapt in my own newly found role as group leader. Secondly, my colleagues and friends Søren M. Karst and Rasmus H. Kirkegaard, whom I have persuaded to join me on further adventures down the rabbit hole! Furthermore, the long list of collaborators over the past years, where I have been fortunate to learn from some best scientists in the world (if you ask me). There are too many to mention, but a special thank goes out to Holger Daims, Michael Wagner, Gene Tyson and Phil Hugenholtz.
Mads Albertsen
Latest posts by Mads Albertsen (see all)
- How to spend 10.000.000 DKK (1.3 mill. EUR) - January 31, 2017
- Welcome to the Albertsen Lab! - January 6, 2017
Pingback: EliteForsk scholarship – failure is a virtue – Albertsen Lab